3.5 The Prescription Drug Approval Process
The question then is, how do all the different medicinal drugs get developed, manufactured, and approved as safe and effective for patients to use?
Investigational New Drug Application
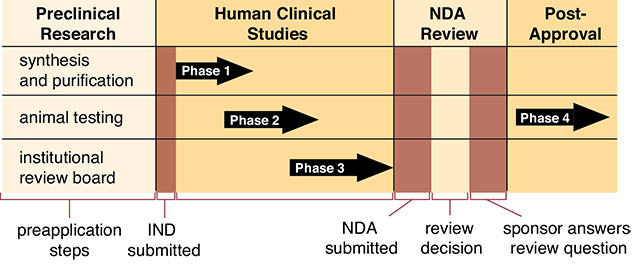
As noted in Chapter 2, a pharmaceutical company must do extensive preclinical laboratory research on animals before an investigational new drug (IND) application can be officially initiated with the FDA. In its application, the developer must offer a compelling case for the human need for the drug, the results of the animal studies, an explanation of how the drug would be manufactured, and a detailed description of the proposed clinical studies on humans.

President John F. Kennedy awarded Frances Kathleen Oldham Kelsey the President’s Award for Distinguished Federal Civilian Service in 1962 for her scientific and ethical stance on thalidomide that saved thousands of children from disfigurement and handicaps. She was later inducted into the National Women’s Hall of Fame.
The research study must then be submitted and approved by the Institutional Review Board (IRB) of the university or hospital where the research will be conducted before the human clinical studies are allowed to begin. The IRB, also known as the Human Use Committee, is comprised of scientists and practitioners from various disciplines, plus consumers. The committee must review, approve, and monitor all medical research involving humans. The proposed studies must meet both scientific and ethical standards to be approved. In most cases each study participant must sign an informed consent form—a document that states the purpose and risks of the research in easily understandable terms.
After the IND is approved by the FDA, non-FDA scientists conduct three phases of human clinical studies as part of the drug approval process.
Phase 1 is the initial study or trial of the new drug, usually completed on a small number of healthy volunteers (typically 20 to 80) over the span of months. The main purpose is to gather sufficient data concerning potential side effects and how the drug is metabolized and excreted. The data are used to design future clinical trials.
The Phase 2 study aims to evaluate the effectiveness as well as the safety of a drug for a specific indication or disease state. Phase 2 studies also involve a small number of patients (typically in the hundreds) and are well controlled and closely monitored by the FDA. Phase 2 usually lasts from several months up to several years.
If the results of Phase 1 and Phase 2 studies are promising, Phase 3 studies are conducted in larger clinical trials (larger groups) to better assess the overall benefits and risks of the investigational drug on patients (see Figure 3.2). Phase 3 studies typically involve thousands of study participants and last one to four years.
Figure 3.2 The FDA Drug Approval Process
New Prescription Drug Application for Brand Name Drugs
If the investigational drug shows promise after Phase 3 studies are completed, the pharmaceutical manufacturer can submit a new drug application (NDA) to the FDA. In the NDA, the applicant must prove the case for both the safety and the effectiveness of the drug before they are permitted to market it. The results of the scientific studies are evaluated by an independent advisory panel of experts, and recommendations are forwarded to the FDA. If the benefit of the drug outweighs the risk, the drug is generally approved. The FDA may request that additional studies be completed. The drug approval process takes a year or longer in most cases. Occasionally, when a medication appears to be very promising early on in testing, the FDA may opt to fast-track the drug and grant early approval. Recent examples of this accelerated process can be seen with the quick approval of drugs to treat patients with HIV or cancer.
Drug development is risky as well as costly. Out of 5,000 to 10,000 screened compounds, only 250 enter preclinical research testing, 5 enter human clinical trials, and 1 new compound is approved by the FDA. The cost of developing a new drug is more than $1 billion, and it can sometimes take as long as 15 to 20 years of research and development to bring a new medication from the laboratory to the pharmacy shelf.
While Phases 1, 2, and 3 of a clinical trial are designed to predict safety of a new drug, serious and unexpected adverse effects can occur after a drug is on the market. For these reasons, a postmarketing safety system is in place, often referred to as Phase 4. Phase 4 requires a drug manufacturer to periodically submit safety data to the FDA. If patients are concerned about safety of drugs, you may want to help them understand the safety checks built into the FDA drug approval process.
Abbreviated Approvals for Generic Drugs
As discussed in Chapter 2, a generic drug contains the same active ingredients as the brand name product and delivers the same amount of medication in the same way and amount of time. Once the patent is due to expire on a brand name drug, a generic pharmaceutical company may submit an abbreviated new drug application to the FDA for approval.
Generic drug applications are termed “abbreviated” because they are not required to include the preclinical (animal) and clinical (human) data to establish safety and effectiveness. Instead, the generic applicants must scientifically demonstrate that their product is bioequivalent, or performs in the same manner as an already approved brand name drug. The drug patent for esomeprazole (sold under the brand name Nexium) expired in 2014.
Currently, esomeprazole is commercially available from several manufacturers who have formulated bioequivalent products. With fewer required studies, generic drugs are far less expensive and require much less time to get to the market.

Concerned over resistance to antibiotics because of overuse, FDA researchers tested the effectiveness of various antibiotics on bacteria in 1978. This same concern continues today among many researchers.
One way scientists demonstrate bioequivalence between brand name and generic drugs is to measure the generic drug’s bioavailability—that is, the rate and extent to which a drug reaches the bloodstream in healthy users and exerts the targeted pharmacological effect. Bioavailability involves how a drug is absorbed, metabolized, distributed, or eliminated from the objective data necessary to compare the generic product with that of the innovator (brand name) drug.
 IN THE REAL WORLD
IN THE REAL WORLD
In October 2015, a new drug designed to boost sexual desire in women was released called Addyi (generic ingredient flibanserin). It exemplifies some of the issues of any new drug. The drug was turned down twice for release by the FDA (as the effects did not seem impressive) and had some specialists petitioning against it. It is now only being released with boxed warnings (warnings that highlight dangerous effects of a drug) and will not be dispensed without patients completing an online questionnaire to show that they understand the potential side effects. This drug is a tablet that must be taken daily for weeks or even months before the positive effects start to manifest. It works on the brain chemicals associated with appetite and moods. However, taking the pill and drinking alcohol can cause extremely low blood pressure and fainting. It has the additional side effects of nausea and dizziness. Also, since it may be prescribed for premenopausal women, the drug effects on pregnancy should be identified and provided to the consumer.
The FDA has developed special regulations for the approval of generic versions of expensive brand-name biotechnology drugs. Unlike for other generic drugs, biosimilars (biologically and therapeutically similar drugs) must demonstrate no clinically meaningful differences in safety and effectiveness when compared to the reference product. The first biosimilar drug was approved by the FDA in 2015, and there were no interchangeable drugs for legal substitution at that point.